Spinal muscular atrophy (SMA)
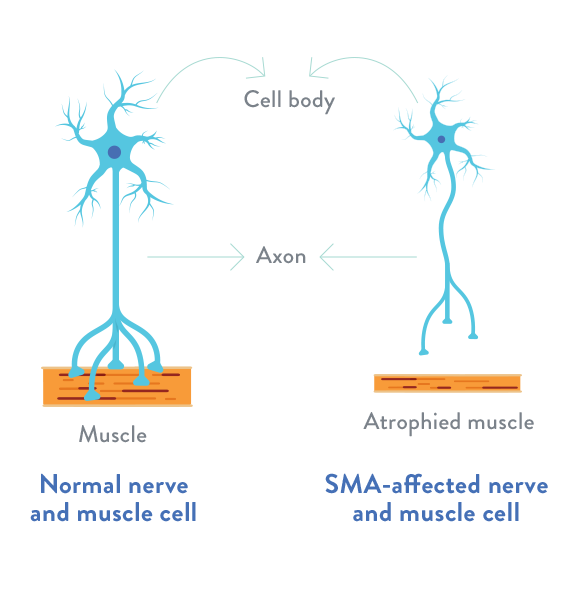
Spinal muscular atrophy (SMA) er alvarlegur taugahrörnunarsjúkdómur þar sem orsökin er dauði á frumum í framhornum mænu sem veldur útbreiddri vöðvarýrnun. Þrjár mismunandi tegundir eru til, í alvarlegasta forminu deyja börnin í æsku. Börn í hinum flokkunum tveimur lifa mun lengur en hreyfifærni þeirra minnkar hægt og bítandi.
Orsök sjúkdómsins
Í líkama okkar er fjöldi gena, sem segja til um ákveðna efnaframleiðslu. Þau eru í svokölluðum litningum sem standa saman í pörum, þar sem annan litninginn höfum við erft frá föður en hinn frá móður. Á litningapari númer fimm er genapar sem er kallað SMN gen. Það segir fyrir um framleiðslu svokallaðs SMN próteins sem er nauðsynlegt taugafrumum í framhorni mænunnar. Þær eru færri hjá þeim sem eru með SMA sjúkdóminn og fækkar smám saman, en mismunandi hratt hjá einstaklingum. Þessar frumur deyja sem sagt vegna skorts á SMN próteininu og það veldur því að taugarnar til vöðvanna verða óvirkar. Vöðvarnir fá því ekki tilætluð skilaboð og taka því ekki á sem skildi. Þeir rýrna svo vegna litlar notkunar. Alger vöntun SMN próteinsins veldur því að allar taugafrumurnar deyja hratt og er það vísun á bráðan bana. Um það bil einn af hverjum fjörutíu einstaklingum hefur annað gen genaparsins gallað. Þar sem eingöngu annað genið er gallað en hitt í lagi kemur það ekki að sök, þar sem hitt genið getur sagt fyrir um efnaframleiðsluna. Hinsvegar ef viðkomandi eignast barn með öðrum, sem einnig hefur annað genanna gallað, þá eru fjórðungslíkur á að barnið hafi bæði genin á genaparinu gölluð. Það eru því einnig fjórðungslíkur á að barnið erfi heilbrigða genið frá báðum foreldrum og beri því ekki sjúkdóminn áfram. Helmings líkur er þá á að barnið beri sjúkdóminn án þess að hafa hann sjálft. Um það bil eitt af hverjum 5.600 börnum sem fæðast hér á Íslandi hafa bæði SMN genin gölluð og eru því haldin sjúkdómnum. Almennt er það þó eitt af hverjum 6.000 börnum fæðist með SMA.
Það vill einstaklingum, sem hafa bæði SMN genin gölluð, til happs að einhverntíma í fyrndinni afritaðist SMN genið og úr varð hið svokallaða SMN2 gen. Vegna ófullkominnar afritunar SMN2 gensins er galli í framleiðsluferli SMN próteinsins, sem veldur því að eingöngu hluti framleiðslunnar er nothæft SMN prótein. Misjafnt er eftir einstaklingum hversu mikið af nothæfu SMN próteini er framleitt eftir forskrift SMN2 gensins, en magn SMN próteins er ákvarðandi fyrir hversu lengi framhornsfrumurnar í mænunni lifa og því einnig hversu þungt sjúkdómurinn leggst á þá sem haldnir eru honum.
Undirflokkar sjúkdómsins
Alvarleiki sjúkdómsins eru mjög einstaklingsbundinn og spannar allt frá því að viðkomandi lætur lífið, skömmu eftir fæðingu, til þess að sjúkdómurinn hefur tiltölulega lítil áhrif á líf viðkomandi. Algengast er þó að sjúkdómurinn hafi veruleg áhrif á líf þeirra sem honum eru haldnir. Þar sem alvarleiki sjúkdómsins spannar jafn breitt bil og raun ber vitni hefur hann verið greindur í fjóra undirflokka.
SMA I – Werding Hoffmann Disease
Greining á börnum með gerð I af SMA er venjulega gerð fyrir 6 mánaða aldur og í flestum tilvikum fyrir 3 mánaða aldur. Þau geta ekki setið óstudd, eiga erfitt með andardrátt og að kyngja. Rúmlega helmingur þeirra einstaklinga sem haldnir eru SMA sjúkdómnum eru með SMA á stigi I og látast þeir flestir fyrir tveggja ára aldur, en aðrir hafa lifað allt fram að unglingsárum.
SMA II
Börn með SMA II sýna einkenni sjúkdómsins uppúr hálfs árs aldri. Þau geta oft setið óstudd þó þau komist ekki óstudd í sitjandi stöðu, einnig geta þau staðið með stuðningi. Öndunarörðugleikar og öndunarfæra sýkingar hrjá gjarnan þessi börn. Þar sem framgangur sjúkdómsins er mjög misjafn getur það verið mjög misjafnt hvað börn með SMA II lifa lengi. Það getur verið mjög snemma eða uppúr 3 ára aldri eða þau lifa fram yfir unglingsár, en þó eru dæmi þess að einstaklingar með SMA II hafi lifað fram á miðjan aldur.
SMA III – Kugelberg-Welander
Börn með SMA III sýna gjarnan einkenni sjúkdómsins upp úr eins árs aldri. Þau geta stað óstudd, en eiga erfitt með gang og þurfa flestir er fram í sækir að nýta sér hjólastól til að komast um. Það getur verið erfitt að standa upp af stól eða beygja sig. Eins og með SMA gerð I og II þá eru öndunarfæra sýkingar það sem ber sérstaklega að varast. Lífslíkur einstaklinga með SMA III eru taldar vera óskertar.
SMA IV
Um er að ræða seinkomið form sjúkdómsins, svokallað fullorðins stig, en einstaklingar með SMA IV sýna yfirleitt ekki einkenni sjúkdómsins fyrr en uppúr 35 ára aldri og framvinda hans er mjög hæg.
Hvað er til ráða?
Engar þekktar meðferðir eru til við SMA sjúkdómnum aðrar en sjúkraþjálfun og heilbrigt og gott matarræði. Þrátt fyrir að sjúkdómurinn sé bæði algengur og alvarlegur hefur allt þar til fyrir fáeinum árum síðan, lítill áhugi verið fyrir rannsóknum sem leitt gætu til meðferðar eða lækningar við honum. Áhugi vísindamanna á rannsóknum tengdum sjúkdómnum hefur þó aukist verulega eftir að genagallinn sem veldur sjúkdómnum fannst árið 1996. Rannsóknir á sjúkdómnum eru fjárfrekar og hefur að mestu verið sinnt af starfsfólki háskóla og ríkisstofnana víða um heim. Alþjóðleg samtök um sjúkdóminn, Families of SMA, hafa að verulegu leyti fjármagnað rannsóknir á sjúkdómnum undanfarin ár. Um er að ræða grasrótarsamtök foreldra og einstaklinga, sem haldnir eru sjúkdómnum. Segja má með sanni að samtökin hafi unnið þrekvirki við kynningu og fjáröflun fyrir rannsóknum. Stærsta einstaka rannsóknin sem samtökin fjármagna í dag er rannsókn Íslenskrar erfðagreiningar á lyfjaefnum, sem gætu gagnast til meðferðar á sjúkdómnum. Meðlimir FSMA á Íslandi hafa tekið virkan þátt í fjáröflun til rannsókna, en alþjóðlegu samtökin séð um ráðstöfun þess fjár. Í dag er raunhæf von að innan fárra ára verði einhverskonar meðferð eða lækning við sjúkdómnum að veruleika. Til þess þarf hinsvegar mikla vinnu og mikið fé.
www.fsma.ci.is